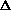 .
The symbol indicates an increase or decrease
of the quantity immediately following.
.
The symbol indicates an increase or decrease
of the quantity immediately following. Charles Thaxton, Walter Bradley, and Roger Olsen have written a book The Mystery of Life's Origin: Reassessing Current Theories. The book was prepared in conjunction with the publishing program of the Foundation for Thought and Ethics, P.O. Box 830721, Richardson, TX 75083-0271, Phone (214) 661-1661, FAX: (214) 661-2989. It was published by Lewis and Stanley, 13612 Midway Rd., Suite 500, Dallas, Texas 75244. Thaxton was academic editor of the creationist text Of Pandas and People.
This review covers the thermodynamics claims in chapters 7, 8, and 9 of MOLO. These chapters are presented on the MoLO web site http://shell3.ba.best.com/~dolphin/mystery/.
The book is sub titled Reassessing Current Theories. The title implies that the authors believe that presently accepted theories regarding evolutionary science have serious shortcomings. However, there is nothing in Chapter 7 to support this contention. Chapter 7, while technically correct, fails in the difficult task of satisfactorily explaining to the lay person the relation between entropy, probability, and the thermodynamics of living systems. It lists numerous references which in general are obtainable only in university libraries and in addition are very difficult for the lay person to understand. The reader is left with the impression, whether true or not, that the authors' approach is more authoritarian than explanatory.
They stress that entropy change results from energy flow, in spite of the fact that a spontaneous entropy change can occur even when no heat flow at all occurs.
The authors state that the probability of thermodynamics change is dictated by the probability of "energy rich" configurations going to "energy poor" configurations, resulting in an increase in entropy. This conclusion is presented using their model of entropy change described as only due to heat flow. This is true in the case of reversible heat flow, but the irreversible case is covered by the use of the greater than or equal sign, as used in their equation 7-9. The significance of the effect of the change in Gibbs free energy is often not mentioned, leading to a discontinuity in the logical development of the mathematical relationships.
Discussion of Mathematics of Chapter 7
In order to understand the mathematical procedures employed by the authors, it is essential to first review the some of the basic mathematics of thermodynamics.
Most thermodynamic equations represent a change in the properties of
a system when it is changed in some manner. Some examples of change are:
(1) A chemical reaction between two interacting systems, as hydrogen and oxygen
combining to form water; (2) Absorption of heat by a system, as when heat
flows into a house during the summer; (3) Absorption of work by a system, as
when air is pumped into a tank; (4) Work done by a system, as when air under
pressure runs an air motor. When these kinds of changes take place, there is
change in the properties of the system. A mixture of two gases becomes a
liquid; the temperature of the house interior rises; the density and pressure
of the air changes. Changes in properties of a system are indicated
mathematically by the Greek letter capital delta: .
The symbol indicates an increase or decrease
of the quantity immediately following.
Internal energy is a property of a system, and a change in internal
energy is denoted as E:
E = q - w (1)
S = q/T (2)
Therefore: E = TS - w (3)
Where: w = work done by the system
q = heat absorbed by the system
E = energy content of the system
S = entropy content of the system
T = absolute temperature
Equation (2) represents the fundamental definition of entropy. Entropy
is a "state function", that is, it depends only on the present
condition of the system. In calculating the change of entropy, it is
generally necessary to determine the amount of heat q absorbed (or evolved,
if there
is a decrease in entropy) when the process takes place reversibly.
(The definition of reversible and irreversible processes will be explained
later.)
In some cases the change in entropy accompanying a process will be the
same, regardless of whether or not the process is
reversible or irreversible. This is the case when there is no work energy
that could be transferred as
a consequence of the change. An example of this would be the heat transfer
when a hot stone is dropped into a bucket of cold water.
However, since both E and S
are fixed for any specific change in the state of the system, equation (3)
is valid only if w represents a fixed value of reversible work energy
transfer.
A clear distinction must be made between the entropy change of a system and the overall entropy change of a system and its surroundings. In a reversible process the entropy change in a system due to the action of the surroundings is equal and opposite in sign to the entropy change of the surroundings. Therefore the overall net entropy change is zero. In an irreversible process the entropy change in the surroundings is greater than the entropy change in the system. Therefore the overall net entropy change is greater than zero.
In many cases the entropy change in a system is
calculated on a theoretical basis as the entropy change in the
surroundings for a reversible process, even though such a process
can not actually be carried out experimentally. In other words, the concept
of reversibility is a mathematical tool. All real processes are irreversible,
although in some cases they may be a very close approximation to reversible
processes. The authors do not always make clear the distinction between
reversible and irreversible processes. Instead they at times merely use
the "greater than or equal" sign (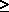 ), as in
their equation 7-9 to cover both reversible and irreversible processes.
), as in
their equation 7-9 to cover both reversible and irreversible processes.
Rearranging equation (3):
TS = E + w (4)
The work done by a system (or sub system) can be a combination of useful work obtained, and work done against atmospheric pressure.
Therefore:
w = PV + w' (5)
Where: w' = work done that can be utilized for useful purposes, eg
lifting a weight or pumping a fluid.
P = atmospheric pressure
V = volume, and V = change in volume
accompanying work against atmospheric pressure.
Combining (4) and (5):
TS = E + PV + w' (6)
If the change is brought about in an irreversible manner so that no work, other than PV work, is done, then w' = 0, and:
TS = E + PV (7)
In this case, since q = TS, the heat evolved (or absorbed) is equal
to E + PV.
The expression
E + PV is called the enthalpy, and
is designated by the symbol H. For processes taking
place at constant pressure without producing (or absorbing) useful work,
H represents the heat evolved or absorbed.
For an enthalpy change at constant pressure:
H = E + PV (8)
S, E, T, P, and V are properties of a system. Referring again to
equation (6), the reversible work w' represents the maximum useful work
that can be obtained from a thermodynamic change. This maximum reversible
work obtainable is designated by G, the Gibbs free energy.
The free energy is a property of the system, like P, V, and T. When work is
done, the free energy content of a system is reduced. For the maximum work
theoretically possible, w' = -G, and from equation (6):
TS = E + PV - G (9)
Rearranging and combining with equation (8):
G = H - TS (10)
and: S = [H - G]/T (11)
If the process is carried irreversibly so that no work is done, for example combustion in a calorimeter, then:
S = H/T
Here, S represents the change in entropy of the
surroundings, not the entropy change of the system. However, the enthalpy
change H depends only on the initial and final
states, and is the same as in the reversible process. Therefore if
S can be determined for a reaction,
G can be calculated from S and
H.
H/T, the irreversible entropy change determined by
combustion in a calorimeter,
is entirely different and often of opposite sign than the reversible entropy
change obtained from the relation
G = H - TS.
NOTE: A positive value (heat absorbed) of
H does not mean the reaction can not take
place. All that is necessary is that the values conform to the equation
G = H - TS.
G can be determined by direct measurement of the
emf of a voltaic cell. It can also be calculated from absolute entropy
values obtained from measurements of heat capacity versus absolute
temperature. The mathematics of thermodynamics permits the calculation
of the effect of pressure and temperature on values of
S, H and G.
The theoretical maximum potential for useful work is represented by
G.
When the value of G calculated from equation (10) is
negative, the change
can occur, although the use of a catalyst may be necessary to make it
happen. Note that it is not always necessary for heat to evolve for a
change to occur spontaneously. H can be zero or even
less than zero.
When H is zero,equation (10) becomes:
G = -TS (H = 0)
An example of H equal to zero is the
free expansion of a perfect gas. The internal energy of the gas is unchanged
on free expansion, and no P-V work is done. In this case there is an
entropy change even though there is no energy flow. If the same change
takes reversibly, with no heat flow (adiabatic conditions), the work output
is equal to G. G is the same
in either case, except that in the reversible process it represents the
work actually obtained, while in the irreversible free expansion process it
represents the work that could have been obtained.
The authors make the statement:
It has often been argued by analogy to water crystallizing to ice that simple monomers may polymerize into complex molecules such as protein and DNA. The analogy is clearly inappropriate, however. The
The authors have previously defined equilibrium as the condition in which the flow of heat is equalized so that there is no temperature differential driving force. Eliminating the time factor, which is irrelevant in thermodynamics, equation (7-9) can be written as:
S - H/T 0 (7-9)
Rearranging equation (10):
S - H/T = - G/T (11)
A reaction can only take place spontaneously when G
is less than 0. In other words, if a change can not produce useful work, it
can not take place spontaneously. If a change is capable of producing
useful work, then its Gibbs free energy will be reduced as a consequence of
that change. Therefore - G will be positive, and if
a reaction takes place spontaneously, then
S - H/T must be greater than
zero. If H is positive, then - H
must be negative, so that S must be positive in order
to satisfy the inequality. The authors have taken a rather roundabout way to
express the simple relation that G must be negative
for a reaction to occur spontaneously. The expression
corresponds to an irreversible change where no work is done.
By contrast the
The statement is misleading in that it does not make clear that the
the fundamental expression for the driving force in thermodynamic
change is G. The problem here is that the other
terms, by themselves, do not provide a reliable basis for the comparison
of one reaction to another.
Referring to equation (10):
In the section, Closed Systems Far from Equilibrium, the authors
treat entropy effects as a manifestation of "entropy flux" due to
"energy flow through the system." They define Se
as entropy flux due to energy flow through the system, and Si
as entropy production inside the system. The total entropy change is
stated as:
S = Se + Si (7-11)
The authors do not provide specific mathematical descriptions of derivations of the terms in the above equation, nor do they provide information on how it could be utilized to determine the free energy of a chemical reaction. With respect to equation (7-11) they state the following:
Where
It would be helpful in understanding Equation (7-11) if the authors would provide some explanation of its derivation. It appears that it is based on the theory that chemical changes in living things can be treated as a flow system:
Energy flow through a system is the equivalent to doing work continuously on the system to maintain it some distance from equilibrium. [snip] An open system offers an additional possibility for ordering-that of maintaining a system far from equilibrium via mass flow through the system, as will be discussed in the next section.Here again, the reader is left to wonder: equilibrium has already been defined as thermal equilibrium. How does that correlate with mass flow, and what exactly is mass flow as applied to chemical changes in living things?
In living plants, the energy flow through the system is supplied principally by solar radiation. In fact, leaves provide relatively large surface areas per unit volume for most plants, allowing them to "capture" the necessary solar energy to maintain themselves far from equilibrium. This solar energy is converted into the necessary useful work (negative
The authors have failed to provide any mathematical support to give credence to their statements. The title of Chapter 7 is Thermodynamics of Living Systems, but there is nothing specific relating to how thermodynamics is actually applied to obtain specific data. Rather the authors summarize the whole chapter as follows:
In mathematical formalism, these concepts may be summarized as follows:
(1) The second law of thermodynamics requires only that the entropy production due to irreversible processes within the system be greater than zero; i.e.,
(2) The maintenance of living systems requires that the energy flow through the system be of sufficient magnitude that the negative entropy production rate (i.e., useful work rate) that results be greater than the rate of dissipation that results from irreversible processes going on with the systems; i.e.,
|(3) The negative entropy generation must be coupled into the system in such a way that the resultant work done is directed toward restoration of the system from disintegration that occurs naturally and is described by the second law of thermodynamics; i.e.,
-where
Concept (1) merely states that the overall entropy must increase for any
real, i.e., irreversible process. With respect to concept 2,
the terms Se
and Si in equations 7-16 and 7-11 are
described respectively as "entropy flux due to energy flow through
the system and entropy
production inside the system due to irreversible processes." Energy
flow is
described as a force that maintains a system far from equilibrium.
No correlation between these concepts and the familiar equations of classical
thermodynamics is given. Concept (3) appears only to state in a qualitative
way that the chemical changes taking place in living things must maintain
the order and complexity of the cell structures of living things.
Perhaps I am missing something, but, at least so far as Chapter 7 is concerned, there seems to be no basis for achieving the goal of reassessing current theories.